La Universidad Pablo de Olavide (UPO) y la Fundación Unicaja han renovado su acuerdo de colaboración para dar continuidad al proyecto BrainCure para BPAN. Esta investigación de vanguardia está dirigida por el prestigioso catedrático de Biología Celular y científico marchenero José Antonio Sánchez Alcázar, cuyo laboratorio en el Centro Andaluz de Biología del Desarrollo (CABD) busca nuevas estrategias terapéuticas contra la neurodegeneración asociada a la proteína beta-hélice, una enfermedad rara ligada al cromosoma X.
El convenio refuerza el apoyo a la biomedicina con potencial traslacional para combatir dolencias de alto impacto social. El equipo del investigador marchenero se centrará en profundizar en los mecanismos moleculares que favorecen la reactivación del cromosoma X inactivo y en evaluar compuestos con actividad epigenética.
El reto de la enfermedad BPAN
La neurodegeneración asociada a la proteína beta-hélice (BPAN) pertenece al grupo de enfermedades raras NBIA, caracterizadas por la acumulación de hierro en el cerebro. Está causada por mutaciones en el gen WDR45, ubicado en el cromosoma X, y provoca:
-
Retraso psicomotor y discapacidad intelectual.
-
Problemas de lenguaje y alteraciones del equilibrio.
-
Convulsiones y deterioro neurológico progresivo.
Hasta la fecha, los tratamientos convencionales como los antioxidantes eran insuficientes para corregir los defectos en el reciclaje celular (autofagia) y la bioenergética mitocondrial de los pacientes.
Una hipótesis innovadora
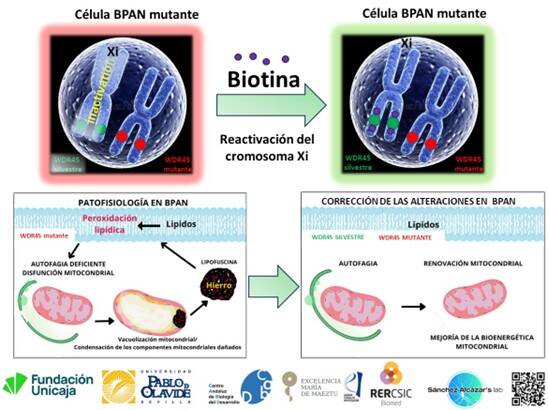
La investigación del grupo liderado por Sánchez Alcázar parte de una premisa novedosa: las pacientes cuentan con una copia mutada del gen, pero también con una copia sana que permanece inactiva en el cromosoma X condensado. El objetivo es utilizar moduladores epigenéticos para 'reactivar' esa copia funcional y corregir los daños.
El grupo ha publicado recientemente un estudio en la revista International Journal of Molecular Sciences donde describen que la suplementación con biotina induce la reactivación del cromosoma X inactivo en modelos celulares de BPAN, corrigiendo las alteraciones fisiopatológicas. Aunque el proceso se encuentra en fase preclínica, combate la raíz del defecto genético sin recurrir a técnicas complejas de edición genómica.
Expansión hacia el síndrome de Rett y el X Frágil
El respaldo de la Fundación Unicaja permitirá al laboratorio del científico de Marchena ampliar los ensayos e identificar nuevos compuestos epigenéticos en células derivadas de pacientes.
Gracias a los resultados actuales, la investigación se ha extendido a otras dos patologías graves del neurodesarrollo ligadas al cromosoma X que carecen de cura: el síndrome de Rett y el síndrome X Frágil. En esta nueva fase, el equipo centrará sus esfuerzos en realizar estudios transcriptómicos, identificar nuevos moduladores y analizar la corrección fisiopatológica en los modelos celulares de las tres enfermedades.